蛋白质测序 (protein sequencing) 是指确定蛋白质(或肽)的氨基酸序列的过程。目的一般是鉴定蛋白质或表征其翻译后修饰。通常,对蛋白质进行部分测序即可获得足够的信息(例如一个或多个序列标签),以便通过与来源于基因概念翻译的蛋白质序列数据库进行比对来识别该蛋白。
蛋白质测序的两种主要直接方法是:
质谱法. 质谱法已成为最广泛应用的蛋白质测序与鉴定技术。
Edman 降解法(Edman degradation)。主要用于表征蛋白质 N 端结构。
多肽从头测序(de novo peptide sequencing)是一种通过串联质谱法鉴定肽氨基酸序列的方法。相比于数据库搜索(database search),从头测序不需要序列数据库。
最初,通过 Edman降解的多肽测序就是从头测序。随着基于数据库搜索的蛋白质组学的流行,“从头测序”的概念反而变得非主流。现在,随着蛋白质组学的发展,根据多肽的串联质谱谱图(MS/MS spectra)中的碎片离子信息直接确定其氨基酸序列变得更容易了,基于质谱的从头测序方法重新引人注目。
解析原理
通过与其中一个氨基酸残基质量相匹配的相同质量差来鉴定序列离子系列(参见表 1)。 例如,an 和 an-1、bn~ 和 bn-1~ , cn 和 cn-1~ 之间的质量差异相同。 鉴定谱图高质量端的 yn-1~-离子。 然后通过将质量差异与氨基酸残基质量进行匹配来继续鉴定 yn-2、yn-3~... 离子(参见表 1)。 寻找已鉴定的 y 离子对应的 b 离子。 b+y 离子的质量是肽的质量+2 Da。 确定 y 离子系列和 b 离子系列后,指定氨基酸序列并检查质量。 另一种方法是先鉴定b离子,然后找到相应的y离子。
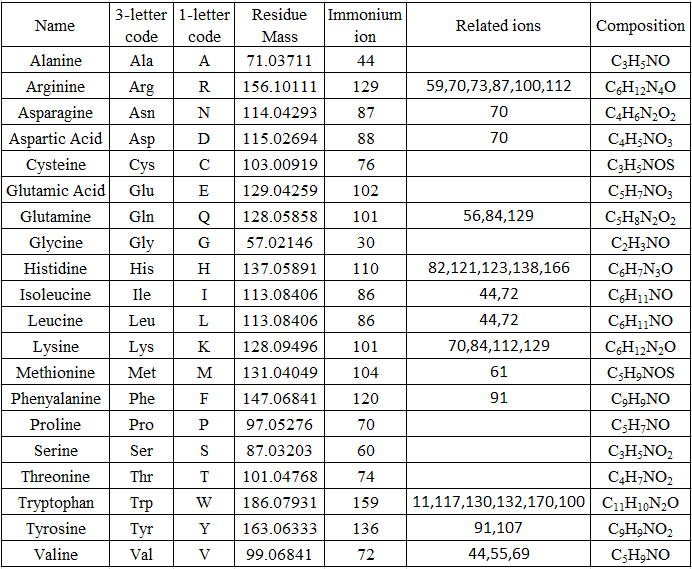
Table 1. Mass of amino acid fragment ions [10.1016/1044-0305(93)87006-X](Huimin Zhong, CC BY-SA 4.0 https://creativecommons.org/licenses/by-sa/4.0, via Wikimedia Commons)
为了便于解释[10.1016/S1044-0305(00)00104-5],b2离子的质量 = 两个氨基酸残基的质量 + 1。

Table 2. Mass of b2-ions in peptide fragmentation (Huimin Zhong, CC BY-SA 4.0 https://creativecommons.org/licenses/by-sa/4.0, via Wikimedia Commons)