软件对定量蛋白质组学研究至关重要。 Cox 和 Mann 开发的 MaxQuant 软件包 [10.1038/nbt.1511] 自 2008 年发布以来,数百项研究已经利用该工具来分析 SILAC 实验。
底层算法和假设通常描述得很差,特别是对于商业软件。 因此,研究人员往往不再手动检查和注释他们的数据,而是或多或少地盲目应用他们最喜欢的软件工具。 与此同时,越来越多的专业出版物报告了对使用单个定量方法生成的数据进行分析的改进,而商业软件中尚未考虑这些方法。
由于基于 MS 的蛋白质组学是一个非常 heterogeneous 的领域,“one solution fits all”的格言很少成立,因此科学家了解数据的性质以及这如何影响适当的计算分析非常重要。
数据处理原理
一级谱图数据预处理通常包括三个步骤:
去除噪声和基线,
谱峰检测,
质量校正和保留时间对齐。
LFQ
常用的软件:
无标记定量方法面临的两大挑战,即共享肽段处理和统计检验。
10.1016/j.mcpro.2023.100581
DIA
计算肽和蛋白质比率
当多个肽段被鉴定时蛋白质的鉴定变得更可靠。每多鉴定一个肽段,蛋白质的定量也变得更可靠(在倍数变化的准确性和精度方面)。
对于蛋白质定量,可以组合对数转换的肽倍数变化来计算对数转换的蛋白质倍数变化。 对数变换对于肽倍数变化分布获得正常形状(即上调和下调对称)是必要的,从而有利于统计分析。
第二个基本假设是测量中不存在任何系统偏差。 这些假设通常并不完全正确。 为了说明在上述假设成立的情况下结合肽对数转换倍数变化(log-transformed fold changes)的有益效果(beneficial effect),请考虑以下情况。 测量一种蛋白质的 N 肽对数转换倍数变化 (pep~ltfc~)。 假设我们还知道肽对数转换倍数变化测量的样本标准差 std(pep~ltfc~)。 如果我们将蛋白质对数转换的倍数变化定义为 N pep~ltfc~ 测量值的平均值,则蛋白质对数转换倍数变化的精度增加将与 肽对数转换倍数变化分布的标准误差成正比。 标准误差等于标准差 std(pep~ltfc~) 除以 N 的平方根,其中N是用于定量蛋白质的肽段数。 由于正态性假设和无偏倚假设并不总是成立,因此蛋白质对数变换倍数变化估计的精度增益将小于这种理想化情况。
肽倍数变化精度和准确度
Peptide fold change precision and accuracy
低强度峰
在所有利用信号强度提取相对/绝对定量信息的技术中,影响肽定量值可靠性的一个参数是用于定量的峰的强度。
与从具有良好信噪比的高强度峰计算出的肽倍数变化相比,从低强度峰(即较差的信噪比)计算出的肽倍数变化必然具有较低的精度,并且更容易产生异常值 [10.1074/mcp.M900628-MCP200]。
对于 MS/MS 谱图的定量,可以通过在较长时间内积累信号来部分补偿样品中肽的相对丰度较低,以改善报告离子的离子统计。 在离子阱仪器中,这可以通过使用自动增益控制来优雅地实现,这确保(理想情况下)在每个肽断裂之前积累恒定数量的前体离子。 不幸的是,三重四极杆或飞行时间仪器的类似机制并不存在。
对于使用 MS1 谱图的稳定同位素方法,通常也不可能提高低丰度肽的肽强度,除非使用 SIM [10.1074/mcp.T400003-MCP200]。 然而,SIM 扫描很少用于发现模式实验,因为它们会显著减慢分析速度 [10.1074/mcp.M500339-MCP200],这对于靶向方法而言不太重要。 幸运的是,通常可以沿着标记和未标记形式的肽的 LC 洗脱曲线获取多个 MS1 测量结果,这可用于提高测量质量。 洗脱曲线尾部的测量明显不如洗脱曲线顶点附近的测量精确,因为这些区域中肽离子的强度低得多。
考虑到上述事实,从单个测量值计算肽段折叠变化的方法的选择并不是微不足道的,通常使用中位数值,因为它相对不太受到低强度峰引入的变化的影响 [10.1038/nbt.1511], [10.1074/mcp.M111.014423]。 对于基于强度的无标记定量,情况完全不同且更加复杂,因为不同条件下肽的强度是在不同的 LC-MS 运行中测量的。 为了计算肽倍数变化,需要确定并比较不同实验中肽的 XIC 顶点的强度或 XIC 的面积 [10.1074/mcp.M700037-MCP200]。
缺失肽
当一个肽在一个实验中被鉴定而在另一个实验中未被鉴定时,必须通过在狭窄的保留时间窗口、小的 m/z 窗口内,并使用同位素分布匹配对齐两个实验来找到“遗漏”肽的 XIC。 如果确实发生不匹配(mismatch),计算出的倍数变化将是错误的,因此在解释此类数据时应格外小心。 建议使用 HPLC-MS 仪器(分辨率、准确度、保留时间稳定性)来最大程度地减少问题 [10.1016/j.immuni.2010.01.013] [10.1074/mcp.M700037-MCP200] [10.1074/mcp.M110.003079]。
“比率压缩(ratio compression)”
在使用同位素标签标记的定量测量中,众所周知的肽比压缩现象限制了可实现的准确度 [10.1021/pr900634c][10.1021/ac201760x]。 在复杂样品中使用“iTRAQ 或 TMT”标记肽测量的倍数变化通常小于真实的倍数变化。 通过分析已知蛋白质倍数变化的样品,这一点已在许多工作中得到验证 [10.1074/mcp.M800029-MCP200], [10.1038/nmeth.1714], [10.1038/nmeth.1716], [10.1021/ac201760x]。
这种效应的物理根源是在选择用于 MS/MS 分析的离子时,其他离子与目标肽的前体离子共同分离。 共分离的离子也会在断裂时产生报告离子,这些离子与目标肽断裂产生的报告离子(带有标识的肽)无法区分。 由于共分离离子的报告离子很可能具有接近 1 的倍数变化(假设实验变化很小),因此它们会将潜在上调或下调的感兴趣肽的测量倍数变化“压缩”至更接近 1。 这种效应可能非常显著,特别是对于低丰度肽/蛋白质。 这个问题可以通过使用窄隔离窗口(即小于 1.5 Th)以及通过使母离子碎片靠近其洗脱曲线的顶点来部分解决 [10.1021/ac201760x]。
尽管仅针对 TMTs 标记描述和研究了压缩效应,但在基于 MS1 的同位素标记定量中,它应该也发生,尽管程度较小。 即使在使用高分辨率质谱仪分析复杂样品时,具有非常相似的 m/z 值的两种不同肽也有不可忽略的可能性会被共洗脱。 这是由于肽分子质量在质量尺度上的分布不均匀 [10.1074/mcp.T400022-MCP200] 以及 ESI 产生多个带电的肽离子,这些离子占据了质谱仪可访问的质荷比(m/z)范围中的相对较小部分。 尚未对此问题进行彻底研究,但应该进行以确定基于 MS1 的定量技术的准确度限制。
信号饱和
当使用最先进的离子检测系统时,signal saturation 已不再是一个问题。
生物学原因
源自同一蛋白质的肽的不同倍数变化可能存在生物学原因,这些变化完全独立于所采用的定量技术。 针对同一蛋白质鉴定的两种不同肽的预期倍数变化应该是相同的。 然而,确实会发生倍数变化在没有明显技术原因的情况下强烈偏离 1 的情况,从而产生异常值。
此类情况可能源自肽的部分 PTM 修饰,或者源自具有截短序列的蛋白质亚型的存在 [10.1038/msb.2012.4]。
正确的蛋白质推断是一个非常重要且经常被忽视的问题。 准确确定应使用哪个肽组来定量蛋白质或蛋白质组并不总是微不足道的。 通过顺序成对组合蛋白质,其中一种蛋白质由属于另一种蛋白质子集的肽来识别(两个肽集也可以相同),可以建立 protein groups [为简洁起见,本文使用“蛋白质”指 protein groups]。 接下来应该优先使用单个蛋白质特有(unique)的肽来定量 protein group [10.1038/nbt.1511]。
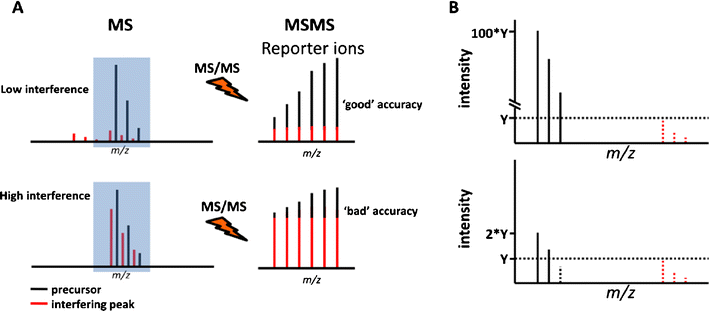
样品复杂性和信号强度对定量精度的影响。 A 定量精度受到信号干扰的限制。 上图:在存在类似 m 的较低强度物质的情况下,在肽质谱中检测到感兴趣的肽(黑线) >/z(红线)。 只要质谱仪的分辨率可以解析这两个物种,就可以从完整的肽质谱中对这两个物种进行定量(基于强度、无标记)。 如果从 MS/MS 谱图(例如,用于绝对和相对定量的同位素标签或串联质量标签)进行定量,则感兴趣的肽将与质谱仪中的干扰物质进行共分离和共片段化 ( 蓝色区域),所得的 MS/MS 谱图包含两个物种产生的报告离子。 在此示例中,共片段化物种的丰度相对较低,仅导致定量准确性的轻微损失。 下图:与上图相同,但显示信号干扰更强的情况。 对于这两个物种来说,基于强度的无标记定量仍然是可能的,但在 MS/MS 谱图中使用报告离子进行定量的准确性将大大降低,导致严重低估真实比率(比率压缩)。 B 定量精度受到信号强度的限制。 上面板中的轻 SILAC 信号具有非常高的信噪比(例如 100,由噪声阈值 Y 定义)。 然而,较重的 SILAC 信号低于噪声,因此数据采集软件将其从频谱中删除。 下图:与上图相同,只是轻SILAC强度为2Y并且重SILAC信号也低于噪声。 在这两种情况下,SILAC 信号比率都是无穷大,这显然不能准确反映真实值,并且往往会导致比率的高估
单肽倍数变化测量
单个肽段定量,特别是对于修饰肽 [10.1126/scisignal.2001570][10.1074/mcp.M110.003079][10.1038/nbt.1759],其精度不如蛋白质定量,原因很简单,蛋白质定量的统计数据多于肽定量。 因此,目前很难很有把握地说特定肽在限定的狭窄范围内具有倍数变化率。
以 SILAC 为例,有人建议,对于磷酸肽,两倍的变化可以被认为是显著的 [10.1126/scisignal.2001570]。 对于蛋白质定量,使用至少三个 SILAC 对达到 1.5 倍的变化时就已经达到显著性 [10.1038/nbt.1511]。 我们敦促读者不要将这些值作为建议,因为它们可能在样本、MS 平台和实验室之间有所不同,而且事实上,可能很难说明变化的重要性。
特别是对于基于强度的无标记定量,人们永远不能完全排除观察到的肽倍数变化是由于 XIC 对不匹配造成的可能性,尽管在使用高端仪器和新的数据比对算法时,发生这种情况的可能性很小 [10.1016/j.immuni.2010.01.013][10.1074/mcp.M700037-MCP200][10.1074/mcp.M110.003079]。
通过谱图计数对单个肽进行定量通常根本没有意义。
使用靶向 SRM 方法通常可以实现更高的单肽定量灵敏度和精度。
进一步需要注意的是 PTM 的定量,根据定义,PTM 是单肽定量。 这里,所确定的带有 PTM 的肽的变化必须通过潜在的蛋白质变化进行校正,以避免实验中蛋白质表达变化引起的误解。
最后,如果单个肽定量很重要,那么变化的显著性通常不应来自单个实验,而是来自重复。
蛋白质倍数变化测定
从前面的讨论可以清楚地看出,我们不能假设所有肽倍数变化都具有相同的质量,并且这会对将它们组合起来计算蛋白质倍数变化产生影响。
处理潜在异常值的一种方法是使用蛋白质的肽倍数变化的中位数 [10.1038/nbt.1511][10.1074/mcp.M111.014423][10.1016/j.immuni.2010.01.013]。 与平均值相比,中值受肽比率分布上限和下限异常值比率的影响要小得多。 无论倍数变化是否进行对数转换,中位数都会给出相同的结果。
或者,可以使用肽对数转换倍数变化的修整平均值。 例如,20% 修剪均值(trimmed mean)意味着在执行计算之前删除对数转换倍数变化的前 10% 和后 10% [10.1074/mcp.M900628-MCP200][10.1021/pr1012784]。 由于较高的强度与较高的精度密切相关,因此人们还可以根据其组成峰强度为不同的倍数变化分配权重。 为低强度离子分配较低的权重将减少它们对整体蛋白质倍数变化的影响。 使用所有数据通常会比断然拒绝低强度峰产生更好的结果,特别是在肽倍数变化统计数据较低(即很少定量的肽)的情况下。 在最简单的形式中,这相当于将一种条件下肽信号的强度相加,然后除以另一种条件下各个信号的总和 [10.1074/mcp.M111.011635][10.1021/pr2008225]。 此过程与两种不同条件下的强度对的引导程序选择相结合,还可以计算蛋白质倍数变化的置信区间 [10.1021/ac201760x]。 在更精细的方法中,可以从大型数据集中“学习”峰值强度和强度变化之间的关系,并用于计算肽倍数变化的权重。 然后将蛋白质对数变换倍数变化计算为加权肽对数变换倍数变化的总和,其中使用的权重总和为 1 [10.1021/pr1012784]。
尝试解决该问题的第一个近似方法是计算所谓的肽倍数变化的提升中值(boosted median of the peptide fold changes)。即,在计算中位数时仅一半数据考虑使用最极端倍数变化 [10.1021/pr1012784]。 尽管这确实对蛋白质倍数变化的准确性产生了微小的积极影响 [10.1021/pr1012784],但需要考虑到不同蛋白质的肽很可能受到比率压缩的不同影响,需要采用更精细的方法来解决这个问题。
再次强调,比率压缩效果可能很强。 一方面,人们可能会认为这会导致对变化的估计相当保守(即实际变化大于数据暗示的变化)。 另一方面,如果某个蛋白质的存在/不存在很重要(例如,在分析敲除体系时),这也可能会产生高度误导。
总之,随着定量的肽数增加,蛋白质的定量结果变得更加精确。 通过分析相同样品的技术重复可以实现非常精确的蛋白质定量 [10.1021/pr1012784][10.1016/j.jasms.2010.01.012]。
我们通常还建议在样品中添加已知倍数变化的蛋白质,以便正确评估复杂样品中的定量准确性 [10.1074/mcp.M900628-MCP200][10.1021/ac201760x]。 对于同位素标签标记,这可以通过以下方式轻松实现:对含有不同标记的蛋白质混合物的低复杂性样品的等分试样进行标记,并以所需比例混合等分试样(例如,Sigma 的 universal protein standard 2 涵盖了大浓度范围内的 48 种蛋白质)。 随后应按原样分析样本,以确认测量的倍数变化与混合比例一致(如此低复杂度的样本不应遭受比率压缩)。 将蛋白质混合样品添加到标记的感兴趣的复合样品中,进行测量,与混合比例相比,添加的蛋白质的倍数变化; 倍数变化的偏差大小将是复杂样本中比率压缩大小的良好指标。
处理异常值时应格外小心。 特别是在定量 PTM 的背景下,异常值可能构成最重要的生物信息,因此不应被断然拒绝。
看不见的离子——处理无穷大
基于 MS 的定量中一个重要但经常被忽视的问题是对一种或多种治疗条件的离子信号缺失的情况的处理。考虑一个简单的对照与处理 SILAC 实验,其中对于给定的肽,我们发现强度为 X 的轻离子形式,但没有相应的重离子,即强度为零。 这意味着该比率是无穷大。 此外,考虑两个这样的 SILAC 对,检测到以高于检测阈值 Y 100倍的强度的一个肽,和强度 2Y 的另一个肽。 在这两种情况下,比率都是无穷大,但两个 SILAC 对的信息内容显然非常不同。 人们可能会忍不住忽视这样的观察结果,但这可能会非常浪费,因为该观察结果可能在生物学上很重要。
大多数情况下,丢失离子的原因很可能不是它们完全缺失,而是信号隐藏在噪声中。 事实上,仪器数据采集软件通常在 MS1(和 MS2)谱图中使用特定的信号截止值来降低数据复杂性。
对于轨道陷阱仪器,一般会丢弃质荷比(m/z)范围内所有检测信号的标准差的 2.4 倍以下的离子 [10.1007/s00216-012-6203-4]。因此,对于高复杂性样品,这个阈值通常会比低复杂性样品更高。这意味着如果轻肽的强度为2Y,其中Y是这个特定MS1扫描的信号截止值,那么我们只能说重肽与轻肽的不可见比率至少为2,这就是这个比率应该报告的方式。 如果强度为100Y,那么我们知道我们至少有100倍的比率。显然,这两种情况在我们获取有关处理效应的信息方面有很大的不同。因此,正确报告这些情况需要报告噪声值Y,这个值可以从Orbitrap谱中轻松提取,特别是在进行个体和/或修饰肽的定量分析时。
方法比较研究
最近有几项研究报告了不同类型的基于 MS 的定量方法 [10.1074/mcp.M111.014423] [10.1021/pr2008225] [10.1021/pr200748h] 之间的详细比较。 我们注意到,此类研究的结果不应无理地概括,因为最终,从样品提取到使用质谱仪进行测量的整个过程对定量研究的整体精度和准确度具有重大影响。 此外,所有最近的比较研究都是使用离子阱/Orbitrap 仪器进行的,因此可能并不总是适用于其他 MS 平台。
引用的工作得出的趋势是同位素标签标记最精确(precise),其次是代谢标记、mTRAQ 标记和其他基于 MS1 的稳定同位素方法,最后是无标记定量。 就准确性(accuracy)而言,代谢标记、化学标记和基于强度的无标记定量表现同样出色,没有任何显著偏差,而同位素标记则表现出系统准确性偏差,但倍数变化率不太明显。
尽管不同的技术都有其优点和缺点,但它们都可以提供高质量的定量数据。 Association of Biomolecular Resource Facilities 最近的一项研究得出了一个非常有趣且有教育意义的结论:定量质量的主要决定因素不是所使用的技术,而是实验室使用技术的经验水平。 因此,比较研究的主要教训是投入足够的时间和精力来掌握一种或两种更好的互补定量技术。
实验中的显著性
独立于所应用的定量方法,在任何研究中检测到的特定蛋白质或肽子集的差异丰度的统计显著性都需要充分的认识。 在只有一小部分蛋白质显示变化的实验中,可以通过分析蛋白质分布的宽度并计算感兴趣的蛋白质位于该分布内的概率来简单地评估。 如果数据遵循正态分布(例如通过 Kolmogorov–Smirnov 检验进行评估),则可以使用简单的显著性检验。 示例包括 t 检验(前提是至少有 3 个重复)、Fisher 精确检验(一次或多次重复)和单向方差分析(一次或多次重复,通常用于 time course 数据分析)。
为了比较谱图计数数据(其原始数据分布类型与使用 MS 强度的方法的数据完全不同),幂律全局误差模型(power law global error model) [10.1074/mcp.M700240-MCP200] 已从微阵列领域改编而来。
具体参见 Neilson 等人的文章 [10.1002/pmic.201000553]。
不同实验间的显著性
尽管在微阵列领域很常见,但公共存储库中发布或保存的大量蛋白质组数据的利用相对较少。 催生新数据分析策略的一个特殊沃土是蛋白质相互作用网络的分析 [10.1038/nmeth.1302], [10.1101/gr.473902], [10.1046/j.1432-1033.2003.03428.x], [10.1038/ncb1086], [10.1038/nature04532], [10.1038/415141a], [10.1073/pnas.0606379103]。
在这些研究中,使用抗体或表位标记的“诱饵”蛋白的亲和纯化 (AP) 与 MS 结合,用于鉴定共纯化的“猎物”蛋白 [10.1021/pr2011632]。
AP-MS 实验中的一个挑战是区分真实的相互作用与高丰度的细胞蛋白质,这些蛋白质通常作为污染物被共纯化,从而描绘出特定的复合物。 为了从 AP-MS 数据推断单个复合物或相互作用网络,需要使用理想数量的诱饵进行大量此类实验。
AP-MS 数据的定量维度(主要是谱图计数或 SILAC)在这里有很大帮助,因为在交替使用所述猎物作为诱饵的实验中以相似丰度水平鉴定的猎物很可能形成独特的蛋白质复合物亚基。 换句话说,如果蛋白质 P1,…,PN 形成复合物,那么在 Pk 为诱饵的实验中,蛋白质 P_i (i ≠ k) 应具有大致相同的丰度。
Sardiu 等人的一项研究利用了这一推理 [10.1021/pr900073d],它使用了几种聚类方法,导致核心复合物及其在蛋白质相互作用网络内各自更远的附着物的分离。
Choi 等人 [10.1038/msb.2010.41] 首先注意到直接的聚类方法并不理想,进一步完善了这种方法,因为在大多数 AP-MS 数据集中,许多猎物蛋白被识别,但尚未进行相应的诱饵实验。
他们提出了一个两步过程,首先根据所有猎物蛋白质的谱图计数数据创建诱饵簇(clusters),然后确定具有相似丰度的嵌套猎物簇。 该小组还开发了一个概率框架,用于根据谱图计数 [10.1038/nmeth.1541] 和无标记 MS 强度数据 [10.1021/pr201185r] 从 AP-MS 数据推断真正的蛋白质相互作用。
已开发出一种称为 C-score 的替代方法,用于从互补的化学蛋白质组学和 AP-MS 数据 [10.1038/nbt.1759] 推断蛋白质复合体成员。 简而言之,作者使用固定化的泛组蛋白脱乙酰酶 (HDAC) 抑制剂(辛二酰苯胺异羟肟酸)从细胞中捕获 HDAC 复合物。
然后将一组已知的 HDAC 抑制剂用于竞争结合模式,结果表明不同的抑制剂对不同的 HDAC 表现出不同的选择性,令人惊讶的是,对不同的 HDAC 复合物表现出不同的选择性。 这些实验中获得的定量蛋白质数据的层次聚类描绘了几种已知和未知的 HDAC 复合物,但其本身不足以以 95% 的置信度识别不同的相互作用因子。
这是通过使用复杂成员作为诱饵进行的有限数量的 AP-MS 实验提供的。 然后将两种类型实验的定量信息合并为 C 分数。 此外,还开发了一种诱饵方法,可以将错误发现率确定为 C 分数的函数,从而可靠地识别蛋白质复合物相互作用因子。
同样,最近应用了三种正交亲和富集方法来确定 BET 溴结构域蛋白的相互作用子 [10.1038/nature10509],并且沿着这些思路的进一步示例应该会出现在未来的文献中。