基因集富集分析(Gene Set Enrichment Analysis,简称 GSEA),也称为功能富集分析(functional enrichment analysis)或通路富集分析(pathway enrichment analysis),是一种在大量基因或蛋白质中发现被过度代表(over-represented)的基因或蛋白质类别的方法,这些类别可能与不同的表型(例如,不同的生长模式或疾病)有关。
背景
转录组学或蛋白质组学等高通量分析技术通常可鉴定到出数千个基因。为了寻找与疾病相关的基因,需要对这组基因进行功能注释,以更好地理解其背后的生物学过程。如果你要对比两种不同细胞类型(如正常细胞与癌细胞)的表达差异,可使用Gene Ontology (GO)富集分析显著上调或下调的差异表达基因,但是这种方法容易遗漏那些未达到显著性标准、但具有重要生物学意义的基因。GSEA则无需预设差异表达的阈值,而是根据基因在两组样本之间的表达差异程度对所有基因进行排序,然后运用统计方法判断某一预定义的基因集是否在排序列表的顶部或底部富集。^[Nature Genetics 2003]^
GESA聚焦于一组先验定义的基因集的表达变化,从而解决了单个基因表达微小变化难以检测的问题。^[Front Genet. 2020. doi: 10.3389/fgene.2020.00654]^
GSEA vs. GO分析
| GSEA(Gene Set Enrichment Analysis) | GO分析(Gene Ontology Enrichment Analysis) |
|---|
| 核心思想 | 检测基因表达数据中先验定义的基因集在排序列表的两端是否富集 | 检测差异表达基因集中是否显著富集于某些GO术语(功能类别) |
| 输入数据 | 所有基因的表达量或差异分析结果(含排序信息,如logFC、p值) | 一组差异表达基因(如DEGs) |
| 是否需要差异分析 | 不依赖事先筛选差异基因;直接用完整排序列表分析 | 需要先筛选出显著差异表达基因 |
| 统计方法 | Kolmogorov–Smirnov-like 富集得分(ES) + permutation test + FDR校正 | 超几何检验 / Fisher’s Exact Test / χ² 检验 + FDR校正 |
| 先验信息来源 | MSigDB、KEGG、Reactome 等基因集数据库 | Gene Ontology(GO)三类注释:BP(生物过程)、MF(分子功能)、CC(细胞组分) |
| 适用场景 | 数据量大但信噪比低、无明确表达阈值时分析更敏感;适合全转录组、全蛋白组 | 通常用于显著差异基因的功能解释,直观、易于理解 |
| 结果解释 | 强调整个基因集在排序中上调/下调趋势 | 强调GO术语中是否含有显著多的目标基因 |
| 常用工具 | GSEA Desktop, GSEApy, clusterProfiler, fgsea 等 | DAVID, clusterProfiler, topGO, g:Profiler, WebGestalt 等 |
分析软件
GSEA作为一种基因集富集分析方法,在处理基因表达数据时,会选取一个或多个功能基因集(例如,将某个 KEGG 通路视为一个基因集)进行分析。
GSEA分析流程是:首先根据基因与表型或样本类别之间的相关性对所有基因进行排序,然后判断每个基因集中的基因是否富集于与表型显著相关的基因列表的顶部或底部,从而判断该基因集中的基因协同变化对表型或处理样本的整体影响。

目前有多种GSEA 软件,它们使用的算法都是相同的。
以下使用官方提供的软件GSEA Desktop^[PNAS 2005] [Nature Genetics 2003]。
1. 安装
软件下载地址: https://www.gsea-msigdb.org/gsea/downloads.jsp
选择对应操作系统版本(Windows / Mac / Linux)进行下载。
GSEA 需要 Java 环境。
2. 准备数据文件
| # | 文件类型 | 文件扩展名 | 内容说明 |
|---|
| 必要 | 表达数据文件 | .gct 或 .txt | 每行一个基因,每列一个样本,含表达值 |
| 必要 | 表型标签文件 | .cls | 说明样本分组(例如对照 vs 处理组) |
| 可选 | 基因集定义文件 | .gmt | 可从 MSigDB 下载 |
| 可选 | 基因注释文件 | .chip | 用于非标准基因命名转换(如探针 → 基因名) |
这些文件都可以用文本编辑器打开,格式示例:
1. .gct 表达矩阵文件格式(制表符分隔):
#1.2
1000 6
Name Description Sample1 Sample2 Sample3 Sample4 Sample5 Sample6
GeneA NA 5.6 5.3 5.8 3.2 3.5 3.4
GeneB NA 7.1 7.3 6.8 2.0 2.3 2.5
...
2. .cls 表型分组文件格式:
6 2 1
# Control Treatment
Control Control Control Treatment Treatment Treatment
我们以官方提供的数据集为例。
打开网页:https://www.gsea-msigdb.org/gsea/datasets.jsp
下载Diabetes数据集(Diabetes_hgu133a.gct、Diabetes_collapsed.gct、Diabetes.cls)
3. 运行软件
打开软件
点击Load data
把三个文件拖到灰色框中,点击Load these files!

点击Run GSEA
Required fields为必填

点击Run
查看结果:Show results folder,这个文件夹里有生成的图片和数据。双击打开**index.html**文件。
可以与官方提供的分析结果进行对比,验证操作是否正确。

报告表示:样本来自 1 和 0 组,每组各有17个生物学重复。23/50 表示共检测了50个基因集,其中23个显著富集。
每组下方显示的富集基因集,表示这些基因集在该组中高度表达。点击HTML页面中的“enrichment results”可以查看每个基因集的详细富集结果。
富集在某个表型下的基因集表格(如NGT,17个样本)
[upl-image-preview uu85f11b2-b164-4f6f-a30e-967c11c22d6c url=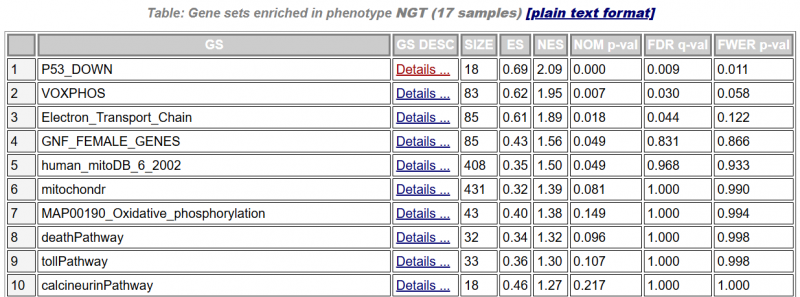 alt={TEXT?}]
alt={TEXT?}]
各列解释如下:
GS:基因集名称
SIZE:该基因集中的基因数量
ES:富集得分(Enrichment Score)
NES:标准化富集得分(Normalized ES)
NOM p-val:名义p值,表示富集结果的可信度
FDR q-val:q值,即多重假设检验校正后的p值
(GSEA通常使用:p值 < 0.05,q值 < 0.25 作为筛选标准)
点击某个条目的 Details 可查看该基因集的详细信息页面:
某基因集的详细统计信息

RANK IN GENE LIST:该基因在排序列表中的位置
4. 结果的可视化与解读
GSEA以两组样本的基因表达数据为输入,识别两组之间的差异表达基因,并按照Fold change对其进行排序,以直观展示各基因在两组中的变化趋势。排序后,位于列表顶部的基因被认为在A组中上调,而底部的则为下调。因此,在GSEA分析中,若某基因集的成员主要集中在排序列表的顶部,则该基因集被认为呈上调趋势;若集中在底部,则为下调趋势(见图A、B)。

GSEA标准输出图形
该图分三部分:

顶部为基因富集得分(Enrichment Score, ES)曲线。该曲线展示了每个基因的累积ES值,横轴为排序后的基因,纵轴为运行中的ES值(Running ES)。曲线的峰值即为该基因集的总体富集得分。
- 若峰值出现在前部,ES > 0,说明该基因集在A组中富集(上调);
- 若峰值出现在尾部,ES < 0,说明该基因集在B组中富集(下调)。
- Leading-edge subset(核心贡献子集):即对富集最有贡献的一部分基因。ES > 0 时,峰值之前的基因为核心子集;ES < 0 时,峰值之后为核心子集。
中部用竖线标记基因集中各基因在排序列表中的位置。若竖线集中在列表前端,说明在A组中富集;若集中于后部,则在B组中富集。
底部展示所有基因在处理前后的变化趋势,通常是Signal2Noise指标的Z分值。图中红色表示在样本A中高表达,蓝色表示在样本B中高表达。
示例:


文献(Free Radical Biology and Medicine 217 (2024) 1–14)中表述:“GSEA implied that the decreased viability in Cry1-silenced KGN cells was mainly attributable to the induction of cellular senescence, ferroptosis, and autophagy, rather than apoptosis (Fig. 2I).”
GSEA算法
GSEA使用的是先验定义的基因集,这些基因集通常是根据其参与的相同生物通路或在染色体上的邻近位置进行分组的。^[PNAS 2005. doi:10.1073/pnas.0506580102]^ 这类预定义基因集可在分子特征数据库(Molecular Signatures Database, MSigDB)中找到。^[Cell Systems. doi:10.1016/j.cels.2015.12.004]^
GSEA 分析的核心是判断某一基因集中大多数基因是否位于基因表达排序列表的极端位置:列表顶部和底部分别代表两种细胞类型表达差异最大的基因。如果一个基因集的基因大多集中在顶部(上调表达)或底部(下调表达),则推测该基因集与两种表型的差异相关。
在标准 GSEA 方法中,分析流程通常包括以下三个步骤^[Nature Genetics 2003]:
计算富集得分(Enrichment Score, ES):该得分表示基因集中基因在排序列表顶部或底部的富集程度,采用类似 Kolmogorov–Smirnov 统计量的方法。
评估ES的统计显著性:通过基于表型标签的置换检验,生成富集得分的零分布,并据此计算 p 值。
这一过程本质上是在检验基因集与所研究表型之间是否存在依赖关系。
多重假设检验校正:在同时分析大量基因集时,需要对多重比较进行调整。各个基因集的富集得分需进行标准化,并计算错误发现率(False Discovery Rate, FDR)以控制错误率。
This can be described as:

Where r is the rank of the gene, p is the power usually set to 1 (if it were 0, it would be equivalent to the Kolmogorov–Smirnov test).
局限性与替代方法
SEA(Simpler Enrichment Analysis)
当 GSEA 于 2003 年首次提出时,其方法学便引发了一些质疑。这些批评促使 GSEA 引入了一些改进措施,包括引入相关性加权的 Kolmogorov–Smirnov 检验、标准化富集得分(Normalized ES)以及FDR的计算,这些要素构成了当前“标准 GSEA”的核心。^[Statistical Methods in Medical Research. 2016]^
然而,GSEA 仍面临一些争议,例如:
为此,提出了一种名为简化富集分析(Simpler Enrichment Analysis, SEA)的方法作为替代。SEA 假设基因之间是相互独立的,并使用更简单的 t 检验方法来进行富集分析。然而,这种简化假设被认为过于理想化,忽略了基因之间的相关性,从而可能导致分析偏差。[Statistical Methods in Medical Research. 2016]
SGSE(Spectral Gene Set Enrichment)
另一个 GSEA 的局限在于其结果高度依赖于用于对基因进行聚类的算法及所设定的聚类数量。[BMC Bioinformatics. 2015]
为克服此问题,提出了一种无监督的替代方法——谱系基因集富集分析(Spectral Gene Set Enrichment, SGSE)。该方法旨在更好地探索 MSigDB 基因集与微阵列数据之间的关联。
其基本分析流程包括两步:
计算主成分(Principal Components, PCs)与基因集之间的关联;
使用加权 Z 方法(weighted Z-method)来评估基因集与数据谱结构(即数据中存在的主要变化方向)之间的总体关联性。
SGSE 强调对数据结构的“频谱式”挖掘,而非对样本间已有分类标签的依赖,因此适用于无监督分析场景。
参考
https://en.wikipedia.org/wiki/Gene_set_enrichment_analysis
https://www.metwarebio.com/gsea-enrichment-analysis-guide/
其他信息:https://zhuanlan.zhihu.com/p/352628317