【预备知识】
- 蛋白质 #55
- 蛋白质的结构由其氨基酸序列决定 #54
蛋白质折叠成能量最低的构象
大多数蛋白质具有特定的三维结构,该结构由蛋白质链中的氨基酸顺序决定。任何多肽链最终的折叠结构,或称构象 (conformation),通常是使其自由能最小化的那一个。
蛋白质折叠的研究始于Anfinsen [DOI: 10.1126/science.181.4096.223],他通过体外实验发现变性蛋白质可以自发恢复其天然结构。用某些溶剂处理(这些溶剂会破坏维持折叠链在一起的非共价相互作用)会使蛋白质解折叠(或称变性,denatures)。这种处理将蛋白质转化为一条失去其天然形状的柔性多肽链。当去除变性溶剂后,蛋白质通常会自发地重新折叠(或称复性,renatures)回其原始构象。由此,提出了著名的热力学假说:氨基酸序列包含了指定蛋白质三维形状所需的所有信息。或者说,蛋白质的一级氨基酸序列决定了其最终的高级结构。
大多数蛋白质会折叠成单一稳定的构象。然而,这种构象是高度动态的,会经历由热能引起的持续波动。此外,当蛋白质与细胞中的其他分子相互作用时,它的构象可能会发生变化。这种形状变化对于蛋白质的功能通常至关重要。
尽管蛋白质链可以无需外部帮助折叠成正确的构象,但被称为分子伴侣 (molecular chaperones) 的特殊蛋白质通常会协助蛋白质折叠。分子伴侣与部分折叠的多肽链结合,帮助它们沿着能量最有利的折叠途径前进。在胞质溶胶拥挤的条件下,需要伴侣来防止新合成蛋白质链中暂时暴露的疏水区域相互结合形成蛋白质聚集体。然而,蛋白质最终的三维形状仍然由其氨基酸序列指定:伴侣只是使达到折叠状态变得更可靠。
α 螺旋和 β 折叠是常见的折叠基序
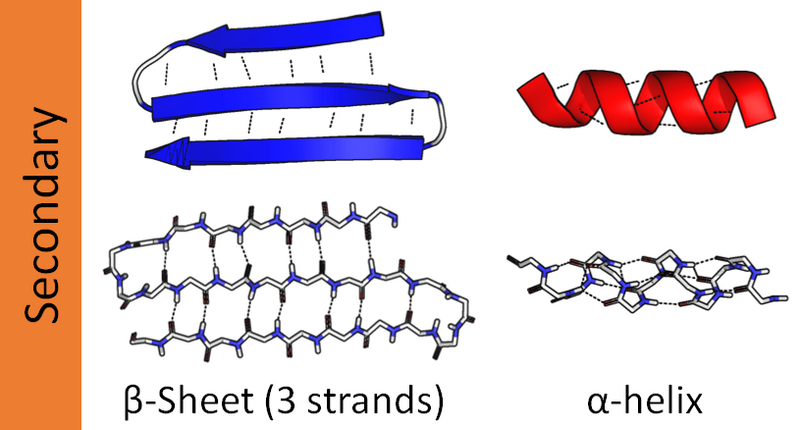
当我们比较许多不同蛋白质分子的三维结构时,会清楚地发现,虽然每种蛋白质的整体构象都是独一无二的,但它们内部经常发现两种有规律的折叠模式。这两种模式是在 70 年前通过对头发和丝绸的研究发现的。
首先被描述的折叠模式称为 α螺旋 (α helix),它是在 α-角蛋白(形成头发中的纤维)中发现的。在发现 α 螺旋的一年内,第二种折叠结构——称为 β折叠 (β sheet)——在丝蛋白(fibroin,丝绸的主要成分)中被发现。
这两种模式很常见,因为它们是由于多肽主链中的 N-H 基团和 C=O 基团之间形成氢键而产生的,不涉及氨基酸的侧链。因此,虽然它们与某些氨基酸侧链不兼容,但许多不同的氨基酸序列都可以形成它们。在每种情况下,蛋白质链都采用一种规律的、重复的构象。在蛋白质的彩带模型中,它们分别由螺旋彩带和一组对齐的箭头表示。
Thomas Shafee, CC BY-SA 4.0, via Wikimedia Commons
许多蛋白质的核心包含大面积的 β 折叠。这些 β 折叠可以由沿着相同方向运行的多肽主链的相邻片段形成(平行链),也可以由来回折叠的多肽主链形成,其中链的每一段运行方向与紧邻的片段相反(反平行链)。这两种类型的 β 折叠都会产生一个非常刚性的结构,由连接相邻链中肽键的氢键固定在一起。
图 《Molecular Biology of the Cell》7E Figure 3–7 两种类型的 β 折叠结构。(A) 反平行 β 折叠。(B) 平行 β 折叠。
图 《Molecular Biology of the Cell》7E Figure3–8 卷曲螺旋 (coiled-coil)。
(A) 单个 α 螺旋,连续的氨基酸侧链以七重序列“abcdefg”进行标记(从上到下)。在这种序列中,位于“a”和“d”位置的氨基酸在圆柱表面上彼此靠近,形成一条缓慢缠绕 α 螺旋的“条纹”(绿色)。形成卷曲螺旋的蛋白质通常在“a”和“d”位置具有非极性氨基酸。
(B) 两个 α 螺旋可以相互缠绕,其中一个 α 螺旋的非极性侧链与另一个的非极性侧链相互作用。
(C) 通过X 射线晶体学确定的卷曲螺旋的原子结构。α 螺旋主链显示为红色,非极性侧链显示为绿色,而亲水性更强的氨基酸侧链(显示为灰色)则暴露在水性环境中。卷曲螺旋也可以由三个 α 螺旋形成。(PDB 代码:3NMD。)
蛋白质结构被认为由四个组织层面构成
将蛋白质的结构依次划分为四个组织层面非常有用。
- 第一个层面是蛋白质的氨基酸序列,被称为一级结构 (primary structure)。
- 多肽链中形成 α 螺旋和 β 折叠的片段构成了蛋白质的二级结构 (secondary structure)。
- 一条多肽链的完整三维组织——包括其 α 螺旋、β 折叠以及在其 N 端和 C 端之间形成的许多扭曲和转折——被称为蛋白质的三级结构 (tertiary structure)。
- 如果一个蛋白质分子是由多条多肽链的复合物形成的,其完整的构象则被称为四级结构 (quaternary structure)。
由于即使是一个小的蛋白质分子也是由成千上万个原子通过精确定向的共价键和非共价键连接而成的,生物学家借助基于计算机的三维显示来帮助观察这些极其复杂的结构。
描述折叠动力学过程的宏观理论
在微观层面,流行的理论包括框架模型 (framework model) 和疏水塌陷模型 (hydrophobic collapse model)。最新的研究倾向于将二者整合为更全面的成核-浓缩模型 (nucleation-condensation model)[10.1016/s0092-8674(02)00620-7]。这些理论加深了对折叠过程的理解,但一级序列如何精确决定高级结构的具体机制仍有待阐明。
研究方向:计算模拟
计算机模拟是揭示蛋白质折叠机制的关键手段。它能在原子和分子水平上,研究水分子、侧链、局部刚性和拓扑结构等因素对折叠热力学和动力学的影响,有望建立一级序列与高级结构之间的确切联系。
然而,目前的模拟研究面临两大瓶颈:
时间尺度限制: 蛋白质折叠通常在毫秒到秒的时间尺度发生,而现有计算机的能力通常只能模拟纳秒到微秒级别的事件。
计算资源耗费与模型简化: 全原子模型模拟计算量巨大,且物理参数过多,难以提取真实的物理意义。因此,研究中通常需要对氨基酸序列进行简化处理。
研究方向:活细胞内的折叠
迄今为止的大多数研究集中在体外折叠。但在细胞内,新生肽链的折叠过程更加复杂:
由于实验技术的限制,人们对细胞内新生肽链的折叠过程知之甚少。但选择性标记 NMR 技术和单分子检测技术的最新发展,使得探测新生肽链折叠过程成为可能[ PubMed]。
未来方向: 进一步利用新技术,研究新生肽链在核糖体上以及受分子伴侣影响的折叠过程,将是蛋白质折叠机理研究的一个重要新方向。
【进阶】
- 蛋白质结构域是构建大型蛋白质的模块化单元 #57
- 利文索尔佯谬 (Levinthal's paradox):如果蛋白质通过随机搜索所有可能的构象来完成折叠,所需时间将远超宇宙的寿命,这与蛋白质在毫秒到秒级完成折叠的现实相悖。